Script and Mantainance
Data processing script
ChIP-seq:
#Mapping single-end $bowtie2 $param -p64 -x $index $fastq | samtools view -C - -T $genome.fa -@ 64 | samtools sort -O bam -@ 64 > $outfile #Mapping paired-end $bowtie2 $param -p64 -x $index -1 $fastq1 -2 $fastq2 | samtools view -C - -T $genome.fa -@ 64 | samtools sort -O bam -@ 64 > $outfile #Quality check ssp $param $single_or_pair -i $input -o $prefix --odir $odir --gt $genometable --mptable $mptable.txt -p 64 >& log/ssp-$prefix #Normalize and generate bigwig parse2wigparam="--gt $gt -i $bam $mp $pair --odir $pdir --outputformat $of -p 64" parse2wig+ $parse2wigparam -o $prefix-raw$mppost-GR -n GR --binsize 100 #Peak Calling macs2 callpeak -t $IP -c $Input -g $species -f BAM -q 0.05 -n $mdir/$peak
ChIA-PET:
mangoscript="Rscript mango_wang.R"
#Non-tagmentation library:
$mangoscript --fastq1 $dir/fastq/${filename}.pair_1.fastq \
--fastq2 $dir/fastq/${filename}.pair_2.fastq \
--prefix $dir/mango/$i --bowtieref $index \
--bedtoolsgenome $genometable \
--chromexclude chrM,chrY --stages 1:5 \
--reportallpairs TRUE --MACS_qvalue 0.05 --threads 50
#Tagmentation library:
$mangoscript --fastq1 $dir/fastq/${filename}.pair_1.fastq \
--fastq2 $dir/fastq/${filename}.pair_2.fastq \
--prefix $dir/mango/$i --bowtieref $index \
--bedtoolsgenome $genometable \
--chromexclude chrM,chrY --stages 1:5 \
--reportallpairs TRUE --MACS_qvalue 0.05 --threads 50 \
--keepempty TRUE \
--shortreads FALSE \
--maxlength 1000
Hi-C, Hi-ChIP:
#Alignment and generate .hic file #docker file is avaible in https://hub.docker.com/r/rnakato/juicer sing="singularity exec --bind /work,/work3 /work/SingularityImages/rnakato_juicer.img" $sing juicer_map.sh $odir hg38 $enzyme $fastq_post -t 60 #Generate contact matrices h1d basic dump $file 25000 all --gt $gt --normalize VC_SQRT -o $name --datatype rawhic --maxchr 22 -n 30 #loops juicer="juicer_tools_1.11.04_jcuda.0.8.jar" java -Xms512m -Xmx200g -jar $juicer hiccups -r 25000 -k VC_SQRT $file $name --threads 12 #TAD juicer="juicer_tools_1.11.04_jcuda.0.8.jar" java -Xms512m -Xmx200g -jar $juicer arrowhead -r 25000 -m 10000 -k VC_SQRT $file $name --threads 24
RNA-seq,etc:
#Mapping with STAR
$STARdir/STAR --genomeLoad NoSharedMemory --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM \
--runThreadN 20 --outSAMattributes All $pzip \
--genomeDir $index_star --readFilesIn $fastq $parstr \
--outFileNamePrefix $odir/$prefix.$build.
#RSEM
$RSEMdir/rsem-calculate-expression $pair --alignments --estimate-rspd --strandedness $strand --no-bam-output -p 40 $odir/${prefix}.$build.Aligned.toTranscriptome.out.bam $index_rsem $odir/$prefix.$build
#edgeR
Rscript $Rdir/edgeR.R -i=$outname.genes.$postfix.txt -n=$n -gname=$gname -o=$outname.genes.$postfix -p=$p -nrowname=2 -ncolskip=1
edgeR.sh rsem/$i/Matrix $db $build $controlnum:$treatnum $controlname:$treatname 0.01
Microarray:
#Use the default setting of GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/)
gset <- getGEO("GSE59908", GSEMatrix =TRUE, AnnotGPL=FALSE)
fit <- lmFit(genemt, design) # fit linear model
cts <- paste(groups[1], groups[2], sep="-")
cont.matrix <- makeContrasts(contrasts=cts, levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2, 0.01)
tT <- topTable(fit2, adjust="fdr", sort.by="B", number=250)
tT <- subset(tT, select=c("ID","adj.P.Val","P.Value","t","B","logFC","GB_ACC","GI","Gene.Symbol","SPOT_ID"))
write.table(tT, file=stdout(), row.names=F, sep="\t")
Maintain and update CohesinDB
We will use two way to maintain (add, modify, delete) the contents included in CohesinDB.
Type 1: The Django admin interface.
We prepared a admin site to easily mantain all contents including:
- Objects for processed NGS file
- Objects for cohesin binding sites
- Objects for cohesin mediated loops
- Objects for cohesin regulated CRMs (genes)
- Other website contents including cell types, update information, etc.
The following shows a list of cohesin site objects in admin interface:
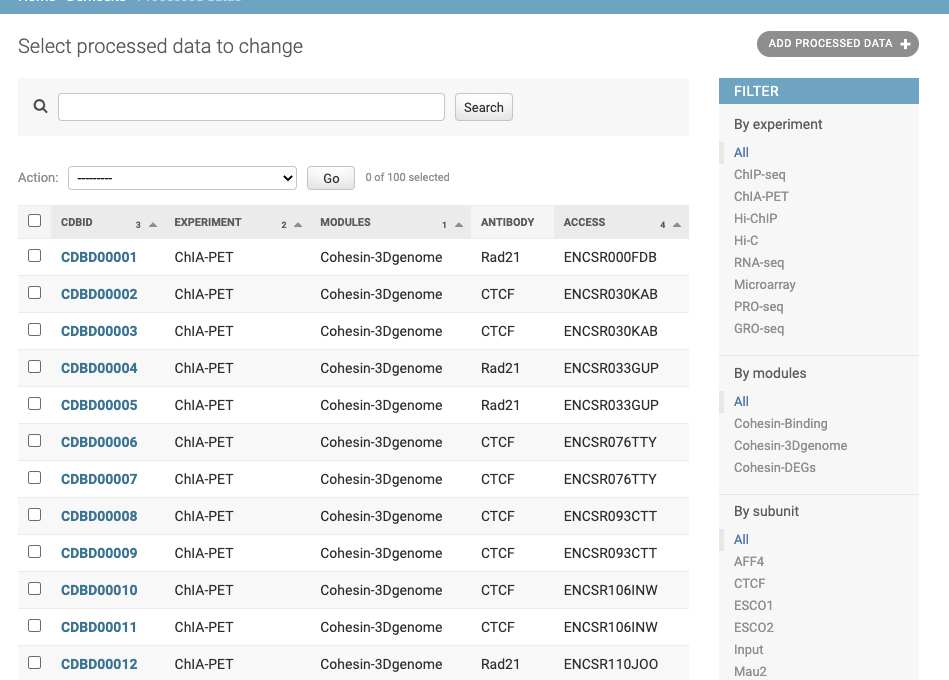
We can delete, add or modify each object for mantainance:
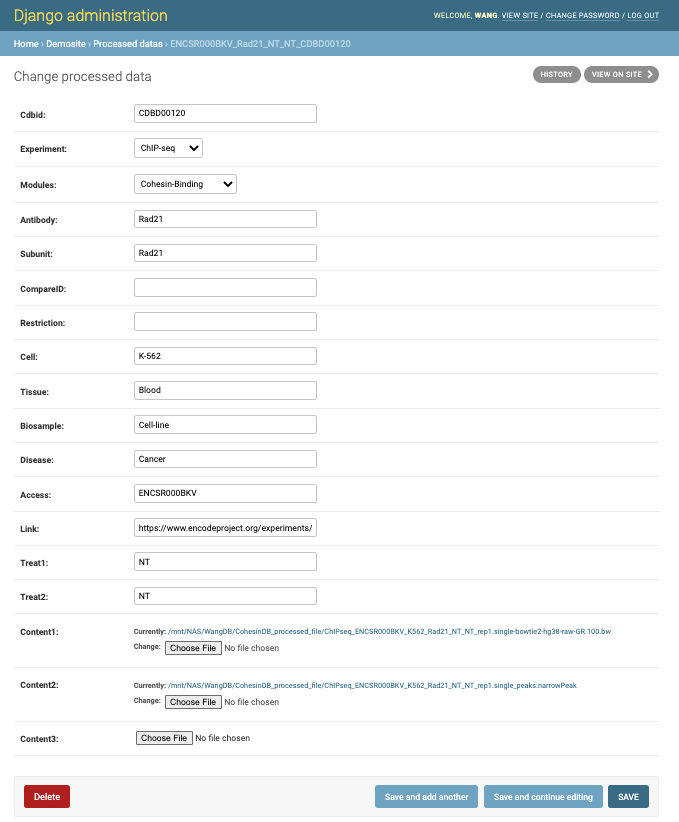
Type 2: Update contents via programmable pipeline.
We have prepared a end-to-end pipeline to update all contents in CohesinDB.
- Process raw sequencing data via the script above
- Generate related objects. Code are open-source in https://github.com/wangjk321/CohesinDB_public/tree/main/makeCohesinObject
- Update CohesinDB content via Django managemant function:
bash makeCohesinSite.sh bash makeCohesinLoop.sh bash makeCohesinCRM.sh bash makeCohesinProcessedfile.sh
#Update Objects for cohesin binding sites python manage.py importPeak #Update Objects for cohesin CRMs (genes) python manage.py importGene python manage.py importCRM #Update Objects for cohesin loops python manage.py importLoop #Update cell type information python manage.py importCell #Update Objects for processed file python manage.py importProcess